Preprocessing Full-Length Single-Cell RNA-Seq for Exon and Junction Read Counts
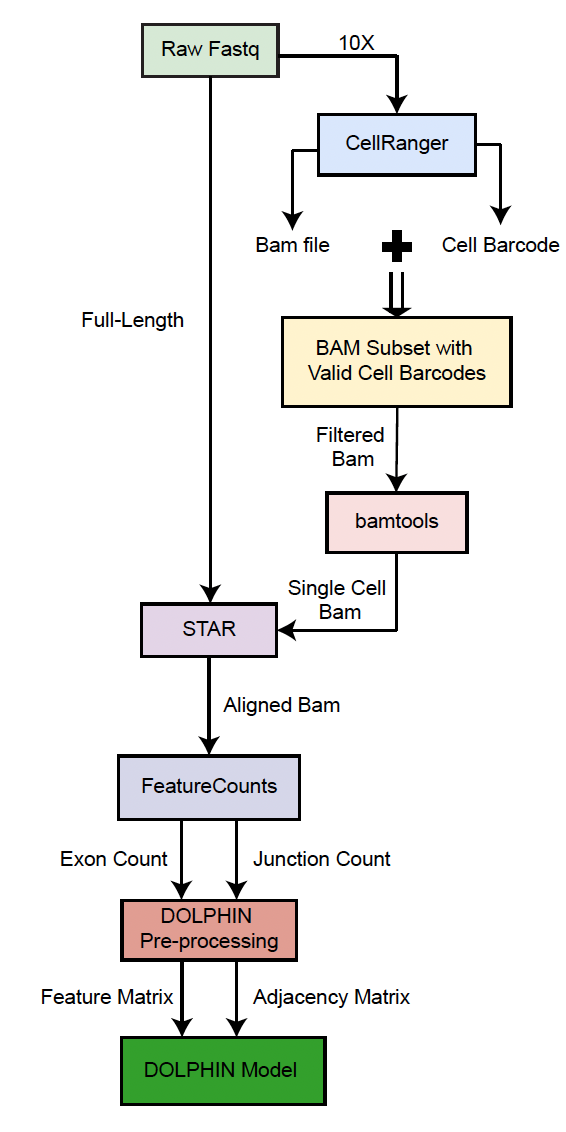
Here is the brief pipeline for full-length and 10x single-cell RNA-seq shown:
Step 1: Download Required Tools
Before starting the alignment process, make sure to download and install the following tools:
STAR >=2.7.3a
featurecounts >=2.0.3
optional: Trimmomatic >=0.39
Step 2: Create a Reference Genome
Run the following command to generate a reference genome for alignment using STAR.
ensembl_mod_indxis the directory where the reference genome index will be stored.Homo_sapiens.GRCh38.dna_sm.primary_assembly.facan be downloaded here.dolphin_exon_gtf.gtfis generated using the file.
STAR --runMode genomeGenerate \
--genomeDir ./ensembl_mod_indx/ \
--genomeFastaFiles Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa \
--sjdbGTFfile ./dolphin_exon_gtf/dolphin_exon_gtf.gtf \
--runThreadN 16
Step 3: Download the Raw RNA-Seq Files
Download the raw RNA-seq files from the provided sources. For the links to the raw data, please refer to the original study.
For full-length single-cell RNA-seq, each cell is stored in a separate FASTQ file. In the following steps, we will process one cell at a time. For example, the codes below processe cell ${ID_SAMPLE}
Step4: Trim
# location of the timmomatic tools
trim = "/mnt/data/kailu/Apps/Trimmomatic-0.39/trimmomatic-0.39.jar"
java -jar $trim SE ${ID_SAMPLE}.fastq.gz ${ID_SAMPLE}.trim.fastq.gz ILLUMINACLIP:/mnt/data/kailu/Apps/Trimmomatic-0.39/adapters/TruSeq3-SE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
Step 5: STAR Alignment
Align to modified exon GTF file and the standard reference genome.
Note: If no gene count table is needed, alignment to the standard reference genome can be skipped.
## `ID_SAMPLE` is the Cell Barcode Name
mkdir ./03_exon_star/${ID_SAMPLE}
STAR --runThreadN 4 \
--genomeDir /mnt/data/kailu/STAR_example/ensembl_mod_indx/ \
--readFilesIn ${ID_SAMPLE}.trim.fastq.gz \
--readFilesCommand gunzip -c \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix ./03_exon_star/${ID_SAMPLE}/${ID_SAMPLE}.
mkdir ./02_exon_std/${ID_SAMPLE}
STAR --runThreadN 4 \
--genomeDir /mnt/data/kailu/STAR_example/ensembl_indx/ \
--readFilesIn ${ID_SAMPLE}.trim.fastq.gz \
--readFilesCommand gunzip -c \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix ./02_exon_std/${ID_SAMPLE}/${ID_SAMPLE}.std.
Step 6: Count Exon Reads and Junction Reads
Get exon gene count using the modified exon GTF file. This will generate the gene count (${ID_SAMPLE}.exongene.count.txt), which will be used later for HVG identification.
mkdir ./04_exon_gene_cnt
featureCounts -t exon -O -M \
-a ./dolphin_exon_gtf/dolphin_exon_gtf.gtf \
-o ./04_exon_gene_cnt/${ID_SAMPLE}.exongene.count.txt \
./03_exon_star/${ID_SAMPLE}/${ID_SAMPLE}.Aligned.sortedByCoord.out.bam
Run the following command to get the exon and junction counts. This step will generate the following files:
${ID_SAMPLE}.exon.count.txt: Exon read counts.${ID_SAMPLE}.exon.count.txt.jcounts: Junction read counts.
mkdir ./05_exon_junct_cnt
featureCounts -t exon -f -O -J -M \
-a ./dolphin_exon_gtf/dolphin_exon_gtf.gtf \
-o ./05_exon_junct_cnt/${ID_SAMPLE}.exon.count.txt \
./03_exon_star/${ID_SAMPLE}/${ID_SAMPLE}.Aligned.sortedByCoord.out.bam